In recent years, artificial intelligence (AI) technology has played a key role in drug design, particularly in predicting Protein-Ligand Binding Affinity (PLA), Drug-Drug Interactions (DDI), compound properties, and compound retrosynthetic design. These applications have accelerated the screening of potential drugs from vast chemical spaces. The team led by Chen Yuqian — Researcher at Peking University Shenzhen Graduate School and Director of the AI for Science (AI4S) Center — has made significant progress in AI-assisted drug design. Their outcome was published inIEEE Transactions on Pattern Analysis and Machine Intelligence(TPAMI, a top-tier AI journal with an IF of 20.8) in December 2024. Related work also includes three papers published inIEEE Transactions on Neural Networks and Learning Systems(TNNLS, a top-tier AI journal with an IF of 10.2) in 2024, and a 2023 study on AI-driven retrosynthetic step inference inNature Communications(NC, a leading interdisciplinary journal with an IF of 14.7).
In current research on protein-ligand binding affinity prediction, deep learning scoring functions based on Interaction Graph Neural Networks (IGNN) have shown excellent performance and development potential. This is largely due to the integration of physicochemical inductive biases into protein-ligand interaction representations in such models, enabling them to learn and capture key interaction features in complex structures for effective geometric affinity prediction. However, many IGNN models still use homogeneous graphs for representation, which partially ignores non-covalent interactions that dominate ligands. In message passing, this approach weakens the impact of covalent bonds within ligand molecules on node feature updates, leading to potential information loss, as shown in Figure 1(a).
Additionally, most 3D-GNN models adopt a "bottleneck" architecture: after multiple graph convolution layers, the input graph structure is typically compressed into a single vector representation via global pooling (as shown in Figure 1(b)). This processing may cause the loss of 3D structural information during compression. To address these issues, the paper proposes an improved affinity scoring method called EHIGN. EHIGN uses a heterogeneous graph modeling approach (Figure 1(c)) and treats binding affinity as the sum of contributions from non-covalent interactions between protein and ligand atoms, with potential biases corrected via a bias correction term (Figure 1(d)). This significantly enhances the model’s generalization ability. Related results were published in TPAMI (https://ieeexplore.ieee.org/abstract/document/10530021).
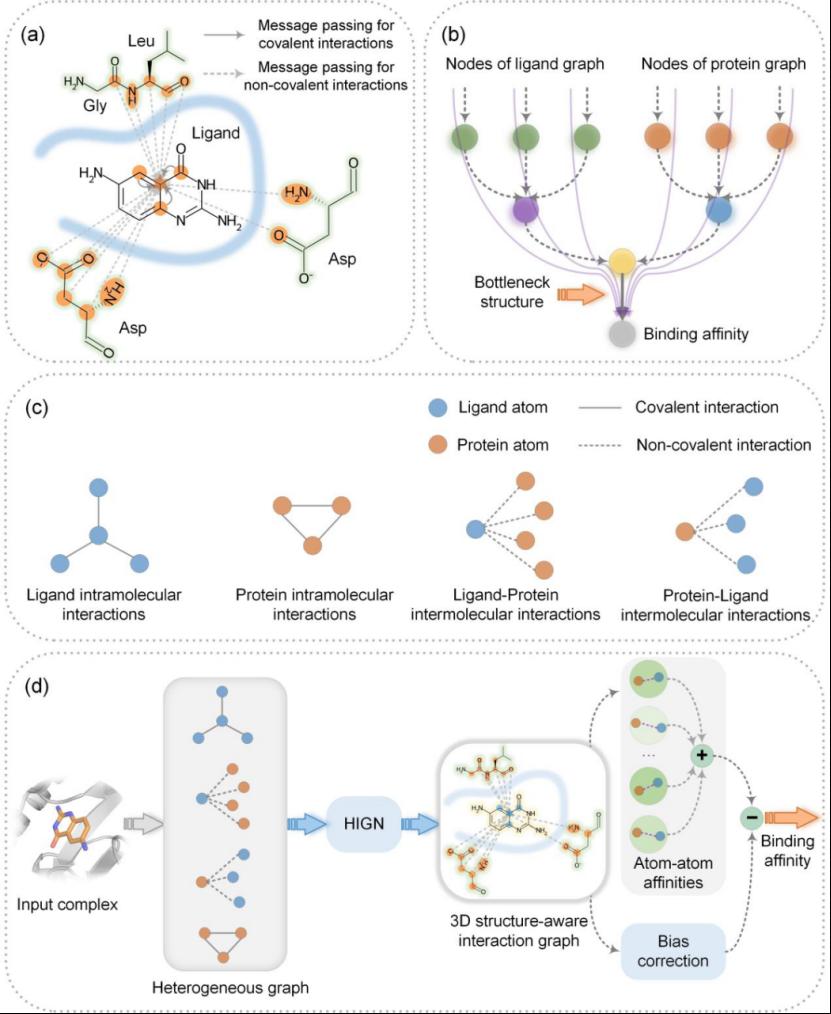
Figure 1 Overall Framework of EHIGN
In the field of drug-drug interaction prediction, although deep learning methods have been widely applied, they still have significant limitations in cross-domain generalization. To address this, the paper proposes a Domain-Invariant Substructure Interaction Learning method for DDI prediction (DSIL-DDI). DSIL-DDI treats substructure interactions as domain-invariant representations of DDI. After extracting substructures using Graph Neural Networks (GNN), a substructure interaction module is used to learn domain-invariant substructure interaction patterns. This module simulates attribute interactions within substructures. To learn domain-invariant representations, DSIL-DDI incorporates an additional loss function to remove noise from irrelevant substructure interactions. For a pair of drugs, the module outputs a DDI representation containing the most critical substructure interaction patterns corresponding to the DDI event. This representation is fed into a classifier to determine the DDI category. For out-of-distribution DDI prediction, DDI representations in unfamiliar domains are computed (without retraining), and these representations are clustered into a specified number of categories. The overall architecture of DSIL-DDI is shown in Figure 2. Related results were published in TNNLS (https://ieeexplore.ieee.org/abstract/document/10044475).
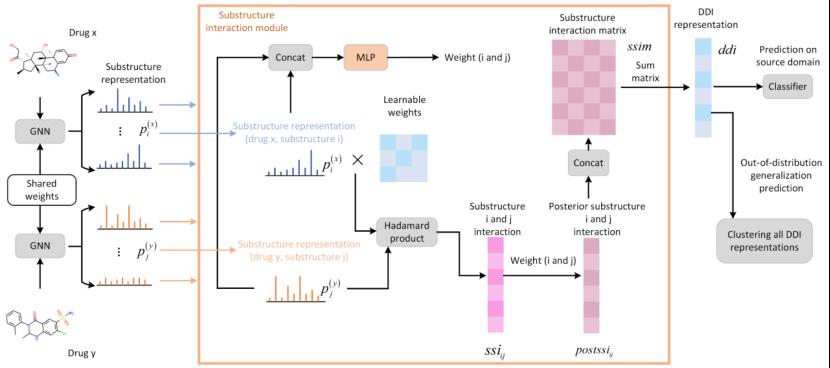
Figure 2 Overall Framework of DSIL-DDI
In compound property prediction, deep neural networks have made significant progress in accelerating and enhancing drug discovery. However, these technologies often require large amounts of labeled data to accurately predict molecular properties. In practice, during the early stages of drug development, the lack of data on physicochemical properties and biological activities of novel molecules or their analogs remains a major challenge. This predicamentmakes applying deep neural networks to few-shot drug discovery extremely difficult.
To address this, the paper proposes a meta-learning framework combining Graph Attention Networks (GAT) — Meta-GAT — for compound property prediction under low-data conditions, as shown in Figure 3. GAT captures local influences of atomic groups at the atomic level through a triple attention mechanism, effectively learning the contribution of atomic groups to overall compound properties. Additionally, the research team constructed a meta-learning benchmark dataset specifically for compound property prediction (Meta-molnet), as shown in Figure 4. Related results were published in TNNLS (https://ieeexplore.ieee.org/abstract/document/10436119;https://ieeexplore.ieee.org/abstract/document/10059171).
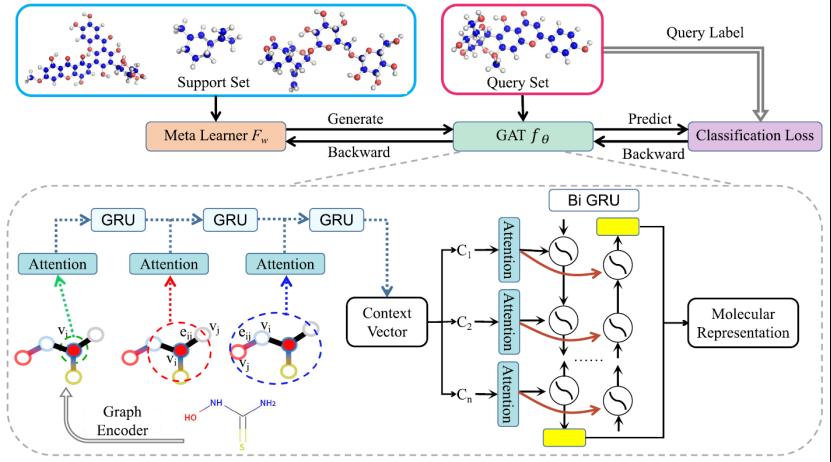
Figure 3 Overall Framework of Meta-GAT
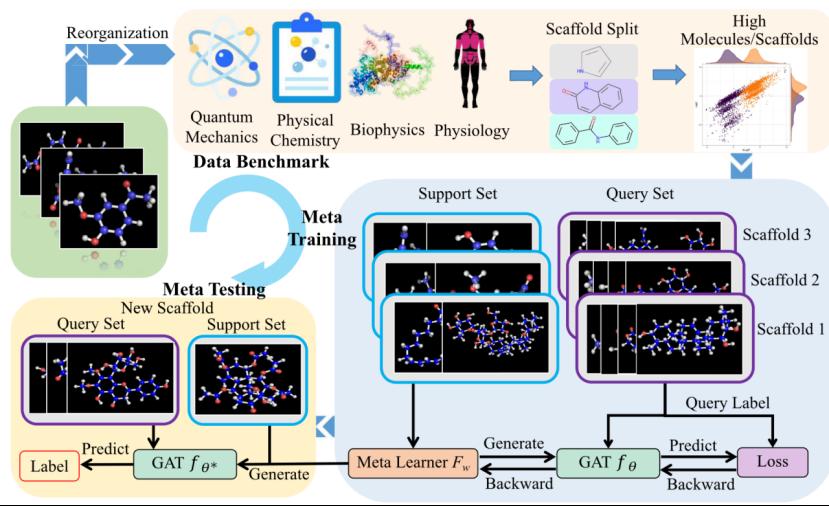
Figure 4 Overall Framework of Meta-molnet
In the field of retrosynthetic optimal step prediction, current retrosynthetic models are limited by their prediction accuracy, diversity, and interpretability, restricting their practical application in synthetic route planning. Enhancing AI-based retrosynthetic prediction models from the perspective of how chemists reason about reactions remains an urgent and important research topic.
To this end, based on the simplified mechanism of reaction transformation, the paper proposes a graph-to-edit architecture called Graph2Edits based on graph neural networks for retrosynthetic prediction, as shown in Figure 5. Specifically, the model frames retrosynthetic reaction prediction as a process of deriving product-intermediate-reactant through a series of interconnected graph edits to learn reaction transformation rules, mimicking how chemists reason about reaction mechanisms. The end-to-end architecture generates graph edit sequences of arbitrary length in an autoregressive manner, strengthening connections between multiple generation steps and improving applicability in multi-center reactions and prediction diversity.
Directional Message Passing Neural Networks (D-MPNN) are used to encode local atom/bond and global graph features, fully leveraging compound structural information to predict atom/bond edits and generate termination symbols. Subgraphs as leaving groups are added to intermediates to complete reactant generation, approximating more realistic reaction transformation processes, significantly reducing generation steps, and improving prediction performance. Related results were published inNature Communications(https://www.nature.com/articles/s41467-023-38851-5).
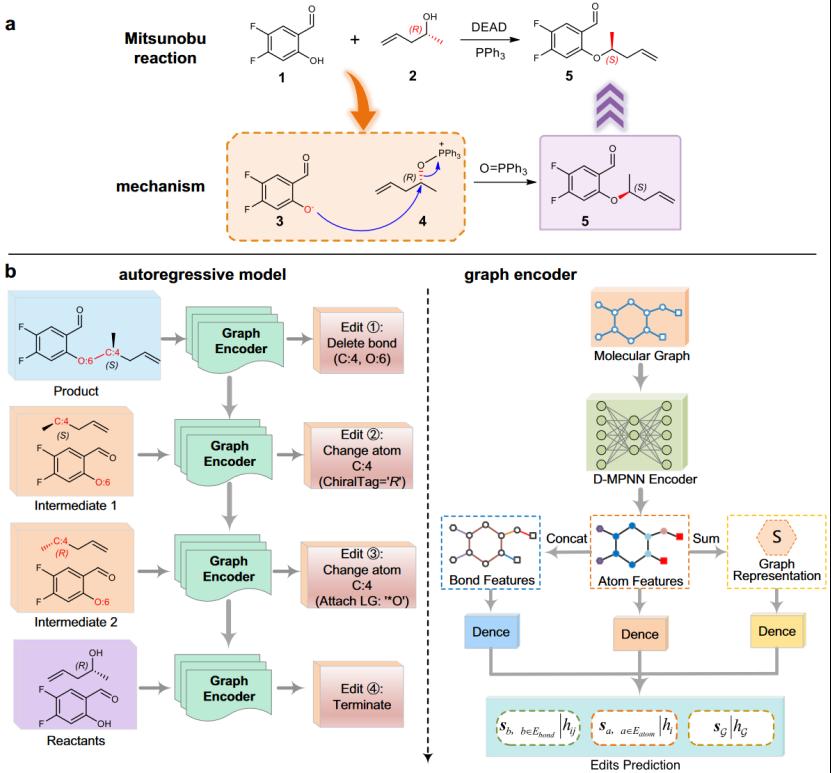
Figure 5 Overall Framework of Graph2Edits

Chen Yuqian is the corresponding author of the aforementioned papers. The research was supported by the General Program of the National Natural Science Foundation of China and projects from Guangzhou and Shenzhen.
References
[1] Z. Yang, W. Zhong, Q. Lv, T. Dong, G. Chen and C. Y. -C. Chen, "Interaction-Based Inductive Bias in Graph Neural Networks: Enhancing Protein-Ligand Binding Affinity Predictions From 3D Structures," in IEEE Transactions on Pattern Analysis and Machine Intelligence, vol. 46, no. 12, pp. 8191-8208, Dec. 2024.
[2] Z. Tang, G. Chen, H. Yang, W. Zhong and C. Y. -C. Chen, "DSIL-DDI: A Domain-Invariant Substructure Interaction Learning for Generalizable Drug–Drug Interaction Prediction," in IEEE Transactions on Neural Networks and Learning Systems, vol. 35, no. 8, pp. 10552-10560, Aug. 2024.
[3] Q. Lv, G. Chen, Z. Yang, W. Zhong and C. Y. -C. Chen, "Meta Learning With Graph Attention Networks for Low-Data Drug Discovery," in IEEE Transactions on Neural Networks and Learning Systems, vol. 35, no. 8, pp. 11218-11230, Aug. 2024.
[4] Q. Lv, G. Chen, Z. Yang, W. Zhong and C. Y. -C. Chen, "Meta-MolNet: A Cross-Domain Benchmark for Few Examples Drug Discovery," in IEEE Transactions on Neural Networks and Learning Systems, doi: 10.1109/TNNLS.2024.3359657.
[5] Zhong W, Yang Z, Chen C Y C. Retrosynthesis prediction using an end-to-end graph generative architecture for molecular graph editing[J]. Nature Communications, 2023, 14(1): 3009.


