The 2024 Nobel Prizes in Physics and Chemistry both went to the field of AI. The former highlighted "how Science is applied and transforms AI", while the latter emphasized "how AI transforms science and people's perception", pushing the research heat of AI for Science (AI4S) to a new high. As it becomes a cutting-edge trend in academia, AI4S is driving a revolution in the scientific research paradigm. The team led by Tian Yonghong and Chen Jie from the School of Information Engineering has been committed to promoting the development of AI4S. Their previous work was shortlisted for the 2022 Gordon Bell Special Prize. Competing on the world stage with teams from Argonne National Laboratory and Oak Ridge National Laboratory in the United States, they stood out among numerous world-class top teams, demonstrating the global top-level capabilities of Chinese artificial intelligence in computing clusters and scientific research innovation. In addition, the team has successively won honors such as the Special Prize for Scientific and Technological Progress of the Guangdong Provincial Science and Technology Award in 2023, the Annual Major Achievement Award of the First "Zu Chongzhi Prize - Frontier Innovation Award in Artificial Intelligence", the First Prize in Guangdong Province and the Second Prize at the National Level in the 2024 "Data Elements ×" Competition organized by the National Data Bureau. On January 17, the team's new AI4S research progress, in collaboration with Researcher Peng Zhou from Guangzhou National Laboratory, was published inNature Machine Intelligence, once again demonstrating the great potential of AI in facilitating the innovation of natural science research paradigms.

Paper link:https://www.nature.com/articles/s42256-024-00966-9
Research Highlights:
(1)This study explored how to customize protein language models for evolutionary prediction tasks, proposed customized pre-training strategies and datasets, and provided a new perspective on the trade-off between protein language model pre-training and downstream tasks.
(2)From the perspective of evolution theory, two fundamental problems of virus evolution were condensed. Through two innovative designs, "weak mutation amplification" and "rare beneficial mutation mining", universal prediction across virus types and strains was achieved, covering SARS-CoV-2, influenza, Zika, and HIV viruses, and realizing a high - degree integration of Science and AI architecture.
(3)The comprehensive reconstruction module of the interaction network where mutations occur (including a dynamic granularity attention mechanism to mine motif patterns) and the proposed multi-task focal loss function are applicable to the general protein system, and have the potential to be further extended for various protein function predictions and protein directed evolution.
(4)Virus evolution predictions at different scales were achieved. In the future, it can be combined with vaccine and protein drug design processes, which is expected to improve design efficiency and controllability.
In nature, species diversity and the proteins that carry functions in organisms are mutually constrained. Proteins, as the carriers of functions, determine the traits of organisms, and these traits, after being screened by selection pressure, result in the current distribution of species diversity. From the perspective of Darwin's evolution theory and continuous new research in epigenetics, biological evolution and the environment form a complex system, representing a co-evolution with the environment. Inspired by this, the research team re-examined the problem of virus evolution prediction from the perspective of evolution theory and proposed a universal evolution prediction model across virus types and strains to solve the two fundamental problems of virus evolution. This model provides a powerful tool for the rapid and proactive update of vaccines and drugs and improves the speed of human response to emerging viral infections, supporting and accelerating the exploration of the complex evolution mechanisms of species.
Mutation is the cornerstone of virus evolution. The specific evolutionary processes of different viruses have their own unique characteristics, but their commonality is that most of the final evolutionary results are almost harmful mutations. From the perspective of the entire evolutionary scale, although the ratio of harmful mutations to beneficial mutations may vary with species and the environment, harmful mutations are always considered to be far more numerous than beneficial mutations. That is, beneficial mutations are an extremely small subset in the evolutionary fitness space of viral proteins. Naturally, the high incidence of harmful mutations makes it difficult for many mutations to coexist within the same variant. That is, the number of mutations in a variant is often small compared to the original type, and only a few sites mutate. Therefore, the team condensed the above virus evolution trajectory into two essential characteristics of virus evolution: "Few-site mutations" and "Rare beneficial mutations". These two evolutionary characteristics lead to obvious modeling challenges: the changes in the intramolecular interaction network caused by "few-site mutations" are relatively weak, making it extremely difficult for neural networks to directly capture them. "Rare beneficial mutations" cause a serious imbalance between positive and negative samples at the data level, making it a huge challenge to accurately predict the rare beneficial mutations that are crucial for evolution.
To address this, the research team proposed the Evolution-driven Virus Variation Driver prediction framework (E2VD) (Figure 1). Through two innovative designs, "weak mutation amplification" and "rare beneficial mutation mining", unified prediction across virus types and strains was achieved. With a customized protein large language model for evolutionary scenarios (trained with the support of 256 NPUs on the domestic E-class intelligent computing platform "Pengcheng Cloud Brain II"), a comprehensive reconstruction module of the interaction network where mutations occur (including a dynamic granularity attention mechanism to mine motif patterns), and the proposed multi-task focal loss function, E2VD achieved the best performance in several key virus evolution driver prediction tasks, significantly and comprehensively outperforming other methods (with performance improvements ranging from 7% to 21%). Experiments have proven that this prediction framework can accurately capture virus evolution patterns, greatly improving the prediction accuracy of rare beneficial mutations from 13% to 80%, achieving a leapfrog improvement in accuracy. It can be flexibly customized and combined to predict evolutionary trends at different scales. It not only realizes the interpretation of the evolutionary trajectory within a pandemic and the accurate prediction of potential high-risk mutations but also achieves the prediction of the macro-evolutionary trajectory at the pandemic scale, reproducing the virus's evolutionary route in the real world and providing theoretical support for interpreting the virus evolution mechanism.
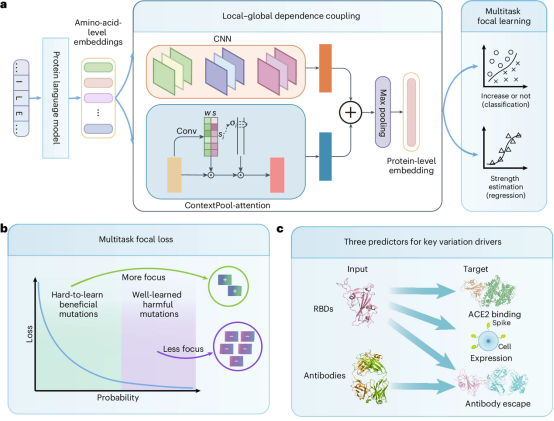
Figure 1: E2VD Model Architecture
In addition, E2VD demonstrated strong generalization capabilities across virus types and strains (Figure 2). The research team proposed a robust evaluation index for changes in virus fitness caused by mutations that avoids the influence of experimental batch effects, and used this to evaluate the generalization performance of the model among different strains of the same virus type and across different virus types. E2VD showed ideal generalization capabilities on SARS-CoV-2, Zika virus, influenza virus, and HIV, consistently outperforming other methods. In the future, it can be further extended to more infectious viruses and combined with vaccine and protein drug design processes, which is expected to improve design efficiency and controllability.
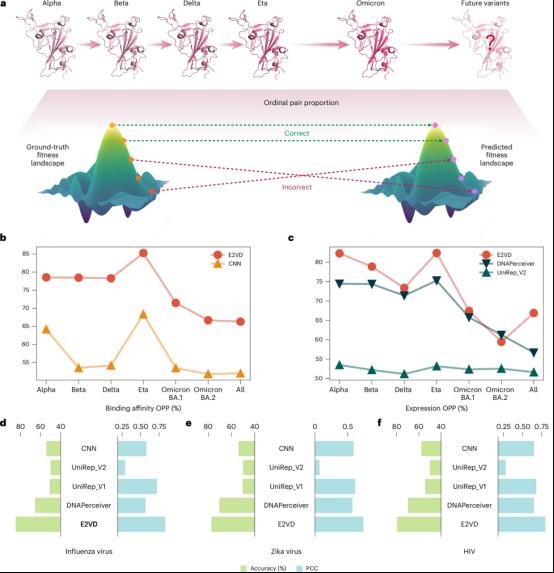
Figure 2: Generalization Performance across Virus Types and Strains
Nie Zhiwei, a Ph.D. student at the School of Information Engineering, Peking University, and Liu Xudong, a master's student, are the co-first authors of this work. Professor Tian Yonghong and Associate Professor Chen Jie are the co-corresponding authors.


